

From Clinical Implications to Modernizing Regulatory Frameworks: Noteworthy Advances To Watch in Cell and Gene Therapy R&D
With a surge in clinical trials, cell and gene therapy research is pioneering treatments, including novel in vivo CAR T options.

Encompassing a broad range of therapies and therapeutic indications, there is never a dull moment in cell and gene therapy (CGT) research and development. Over the last decade, CGT clinical trials have more than tripled, with 76 new therapies launched globally by late 2023.
Given the rapidity of scientifically complex innovations in the space, there are many unknowns associated with CGTs, especially regarding safety and patient access due to specialized manufacturing and administration needs in some cases, complicated logistics and cost of development. However, the uncertainties have not deterred stakeholder interest in evaluating the promise of these novel treatments. For oncology alone, global investment in CGTs is estimated to grow from $5 billion currently to $14 billion by 2029.
Because advances in CGT R&D are plentiful, there is a need to ensure viable options reach patients quicker without compromising safety or quality. This article discusses several points of progress in CGT R&D worth watching with caution for the efforts behind each to better address patient needs.
In vivo CAR T: Reducing logistical complexities while improving scalability of treatment
Much of the clinical trial expansion in CGTs can be attributed to chimeric antigen receptor (CAR) T-cell therapies, specifically traditional ex vivo CAR Ts. Currently available in the US to treat various blood cancers and multiple myeloma, ex vivo CAR T-cell therapies are offering much-needed hope to patients as highly individualized therapies using patients’ own T cells or those of healthy donors, with potential for long-term remission.
While the demand for ex vivo CAR T therapy is high due to the level of personalization, this class of innovative therapies also involves complex, time intensive and costly manufacturing processes and the need for highly precise handling of the treatment from collection through infusion. And because the treatment can only be administered at accredited cancer centers, patient access and related payer reimbursement requirements add to the challenges of real-world use.
To help combat some of those key concerns, drug developers are evaluating in vivo CAR T-cell therapy, which is designed to reprogram a patient’s own T cells directly within their body, bypassing the need for external cell manipulation. This involves administering gene delivery vehicles – such as lipid nanoparticles (LNPs) or viral vectors – that carry genetic instructions for CARs. These vectors target circulating T cells, enabling them to express CARs that recognize and attack disease-specific antigens. Once modified, the CAR T cells expand and activate in situ, initiating a targeted immune response against cancerous or autoreactive cells. This method is faster, scalable and less costly than traditional ex vivo CAR T therapies.
Additionally, advanced platforms for nanoparticles are available for use under Good Manufacturing Practice regulations. This allows large-scale production of in vivo CAR T cells, because they can be stored and kept stable via lyophilized reagents that allow freeze dried cells to be kept at room temperature longer, which can expand patient access.
Though in early evaluation, findings from recent preclinical studies examining in vivo CAR T therapy for treatment of blood cancers demonstrated promising antitumor activity. Researchers are cautiously optimistic about in vivo T-cell engineering, pointing to the potential for off-target delivery of the CAR transgene to other immune cells, triggering adverse responses.
They are also exploring challenges of gene delivery methods, such as an adeno-associated virus vector, while drug developers have to also consider regulatory complexities and concerns about nanoparticle delivery systems. It will be key to monitor limitations in maintaining long-term CAR expression on T cells.
In aiming to better connect the promise of innovative science and actual patient access and reduced logistical overhang, in vivo CAR T R&D is worth watching for additional learnings and insights to gauge its real-world potential. Global pharmaceutical companies’ entry into the in vivo CAR T space is a notable indication of the heightened interest in its potential.
In June, AbbVie acquired Capstan Therapeutics, a clinical-stage biotech company specializing in in vivo RNA technologies, for $2.1 billion. The acquisition includes the biotech’s potential first-in-class in vivo targeted LNP anti-CD19 CAR T therapy candidate in Phase 1 development for the treatment of B cell-mediated autoimmune diseases. More recently, Kite, a Gilead Company, acquired Interius BioTherapeutics and its in vivo platform for $350 million.
RNA therapeutics: Enhancing platform design for ease of therapeutic pivoting
The interest in ribonucleic acid therapies has surged in recent years, specifically following the evaluation, approval and dissemination of the messenger RNA-based COVID-19 vaccine. Between 2020 and 2021, RNA-based pharmaceutical companies grew in the market, topping $228 billion, compared to nearly $3 billion only 10 years ago.
Advances in this groundbreaking field hold promise for rethinking how we treat a wide array of diseases by using RNA molecules as genetic messengers to carry out vital cellular functions in patients’ bodies. CGT stakeholders are excited about the potential to use the platform technology to pivot clinical applications to help address a range of diseases that in some cases are extremely difficult to treat. As seen in Table 1, this includes genetic disorders, metabolic disorders and cancers.
Table 1: Approved RNA drugs by modality and indication.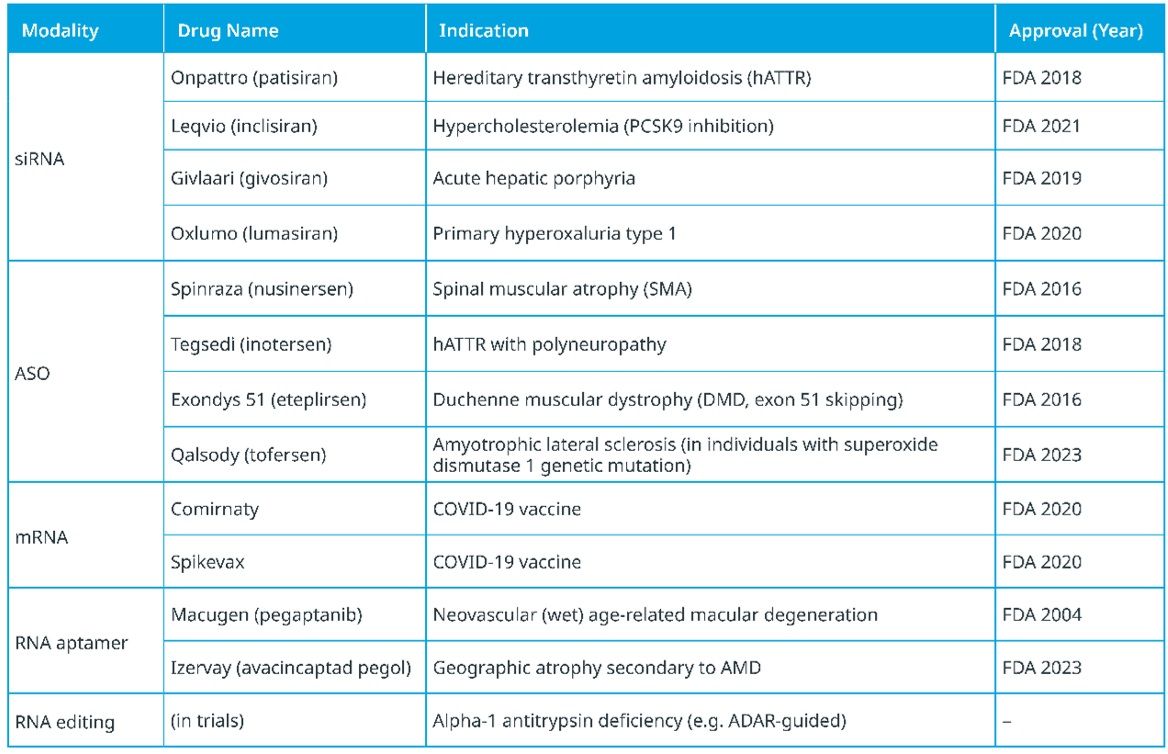
Credit: IQVIA.
In theory, these therapies can target any gene of interest by selecting the appropriate nucleotide sequence on the target RNA. Because RNAs can selectively act on proteins, transcripts and genes to widen the range of druggable targets, it opens up tremendous potential to treat a multitude of diseases. In contrast, currently approved protein-targeting therapeutics (e.g., small molecules and antibody drugs) target only .05% of the human genome. This is due to various factors, including that most DNA sequences of the human genome are transcribed into noncoding transcripts and that approximately 85% of proteins lack specificity in areas for small molecule binding.
Because RNA therapies can be quickly synthesized, once they’ve been identified, candidates can easily be produced at large scale and tested using advances in platform technologies. With sequence editing becoming more straightforward via computational methodologies, the production of RNA-based therapies can accelerate and offer flexibility based on desired target. This ease of development and production makes RNA therapies strong candidates for multi-indication applications.
Recognizing unique nuances in clinical evaluation of oligonucleotide therapies, including RNAs, the US Food and Drug Administration (FDA) issued guidance last year emphasizing several key considerations in trial design. For one, in gauging immunogenicity risk of RNAs, the FDA recommends accounting for patient characteristics. For example, their potential immune response levels, if they have an autoimmune condition, is critical to note, or, if they are taking immunosuppressants like chemotherapy, it may affect therapeutic impact.
Additionally, it is recommended for trial sponsors to use early pharmacokinetic and pharmacodynamic characterization to inform late-phase enrollment with patients with a full range of liver and/or kidney function. Trial sponsors are requested to provide reasoning if patients with impaired renal or hepatic function are ultimately excluded from late-phase trials.
Matching CGT innovation with a modernized regulatory framework
The science behind game-changing innovations in CGTs and others continues to be gathered, evaluated and pushed forward for development. It is equally necessary to emphasize how stakeholder collaboration and ongoing discussions can help drive the availability and use of these novel treatments among those most in need.
The FDA has recently launched a “national listening tour” to initiate dialogue with pharmaceutical and biotech company leadership on how to better support modernized drug development through enhancements within the regulatory framework to complement novel CGT R&D. Rethinking the infrastructure for CGT reviews specifically, the FDA is taking in ideas for increasing both process transparency and flexibility in clinical trial design to accelerate approval pathways for novel treatments.

Achieving High-Throughput CE-SDS While Preserving Data Integrity
This case study outlines a validated approach for implementing high-throughput CE-SDS while maintaining existing SOPs and regulatory compliance across a global organization.View App Note / Case Study
Sharing decision-making rationale
For one, the FDA is working to increase transparency among R&D stakeholders in a way not previously done. In July, the agency published hundreds of complete response letters (CRLs) and/or decisions regarding Investigational New Drug Applications, Biologics License Applications and other submissions from 2020 to 2024 so drug developers can review and understand the agency’s decisions and related rationale. Generally, some of the reasons most cited for issuing CRLs include safety and efficacy concerns, manufacturing deficiencies and bioequivalence issues.
It is becoming clear that beyond safety and efficacy concerns, CGT drug developers need to provide regulators with a comprehensive chemistry, manufacturing and controls strategy, including insights to process validation and readiness.
By reviewing CRLs, CGT trial sponsors can better gauge strategic direction of clinical trial programs and individual trials and related submission strategies. For emerging biopharmas that generate the majority of CGT innovations, there is little room for error, making these insights tremendously valuable for ensuring plans align with regulatory expectations.
Offering flexibility in CGT reviews and minimizing delays
In recent years, the FDA has taken noticeable steps to address delays in CGT reviews due to the unique nature of therapies and technologies. This includes its INitial Targeted Engagement for Regulatory Advice on CBER/CDER ProducTs, or INTERACT program, allowing CGT trial sponsors to seek FDA feedback before pivotal nonclinical studies or filing Investigational New Drug applications. This can help sponsors navigate potential pitfalls, such as complexities within manufacturing processes, early on. The FDA also created the Office of Therapeutic Products within the Center for Biologics Evaluation and Research in 2023 to increase review capacity with additional staff with extensive CGT expertise.
On the other end of CGT R&D, per FDA and European Medicines Agency requirements, CGT trial sponsors are required to conduct long-term follow-up (LTFU) studies for up to 15 years post-therapy to monitor long-term safety. Though the FDA recognizes the unique safety concerns of CGTs, the agency also acknowledges the unique burdens of safety monitoring for multiple years. This is especially difficult when the need is to closely monitor patients over years and related life changes while ensuring quality data collection but limiting the burden on companies and trial sites. Currently, stakeholders are waiting to see whether agency leadership will offer flexibility in CGT LTFU requirements and/or invite input from pharmaceutical and biotech companies.
Additionally, to help accelerate the review of some CGT applications, in 2024, the FDA developed and issued draft guidance for developers regarding its new Platform Technology Designation program. This is intended to recognize purposeful platforms that can be tailored to treat varying groups of patient populations with different diseases and applied to future CGT applications. Developers would be required to show evidence that the platform could help accelerate and streamline complex therapy development and regulatory review without compromising safety or quality. This designation can be especially beneficial to some gene therapies designed to treat very small subsets of patient populations with rare diseases.
In June, the FDA granted the platform technology designation to Sarepta Therapeutics’ rAAVrh74 viral vector used in the investigational gene therapy SRP-9003 (bidridistrogene xeboparvovec) for the treatment of limb-girdle muscular dystrophy type 2E/R4, and within a month, the agency revoked the designation and suspended distribution of the investigational gene therapy citing “three deaths potentially related to these products” and potential patient exposure to “significant risk of illness or injury.” Though not the desired outcome for the first-ever platform technology designation, since patient safety is the ultimate priority, it does emphasize the agency’s efforts to help update CGT review processes for potentially quicker treatment access for those in need.
Cautiously hopeful in a rapidly evolving therapeutic space
While there is much to navigate in CGT R&D, it is an exciting time to watch as scientific advances expand and stakeholders are able to monitor next-generation therapies for the potential they offer patients in need. This is especially true for those facing limited options due to rare diseases or experiencing difficulties accessing novel therapies.
With no standstill of incoming CGT innovation, it will be a short wait to see how transformative therapies are being evaluated from practical, real-world use perspectives by drug developers, regulators, patient advocates and others to create a more favorable CGT ecosystem for patients.




